F I N D G E N E
Description and data preparation
INTRODUCTION *
What is FINDGENE ? *
What is FINDGENE's history ? *
BACKGROUND TO THE METHOD *
DATA PREPARATION *
A note on the implicit animal model. *
Information required for a FINDGENE analysis: *
Pedigree information in a pedigree file: *
Data in a data file: *
Model and parameters. *
FINDGENE results. *
REFERENCES *
CONDITIONS and COSTS FOR FINDGENE ANALYSIS *
CONTACT *
FINDGENE is a computationally fast program developed to detect segregation at an unknown quantitative trait locus (QTL) influencing commercial traits. In the past many genes of significant effect, such as the Booroola gene, have been discovered through simply ``eyeballing'' the data. FINDGENE is meant to facilitate this process for organisations implementing routine genetic evaluations and for researchers who require a ``quick fish'' approach to QTL detection.
FINDGENE will give an indication of the size of any putative QTL and its frequency in the population. In addition FINDGENE will present probabilities, for each animal, of belonging to each of the three major locus genotypes. Identified families, in which the major gene may be segregating, can then be analysed more intensely to confirm the presence of such genes. ``Second string'' approaches may involve iterative sampling techniques, which are very computing intensive.
FINDGENE has evolved over a period of 4 to 5 years. Brian Kinghorn wrote the first version which he used to generate results given in Kinghorn et al. (1993). Improvements were then made by Johan van Arendonk, Margaret Mackinnon and Gerry Davis together with Brian. These improvements included: Johan's methodology for calculation of genotype probabilities (see van Arendonk et al. 1989); implementing a reduced animal model; fitting fixed effects; and correcting for bias in the intercept. This version was designed to be launched from within Microsoft's QuickBASIC environment, giving graphic output of data and posterior distributions.
Following the launch in 1993 of the UNE/CSIRO Findgene project, funded by Australia's Meat research Corporation, Richard Kerr re-wrote FINDGENE completely. Solutions in the regression step were now obtained using an implicit representation of the mixed model equations. A new subroutine was also written to calculate genotype probabilities. The ability of the procedure to handle large data sets was significantly improved, as well as overall speed and robustness.
However problems of bias in estimation of the major gene parameters still persisted. This was finally solved when we were successfully able to correct for bias within each BLUP iterate, as opposed to within each FINDGENE iterate. All estimates of major gene parameters are at least close to unbiased as determined by sensitivity analyses. Finally maternal polygenic effects (EBVs) were included. Fitting these effects are important when analysing such traits as birth and weaning weight.
FINDGENE is a two step procedure, and is described in detail by Kinghorn et al. (1993). A brief description follows. Initial estimates of the effects of a putative QTL in heterozygote and homozygote form are given as well as other priors of gene frequency and polygenic heritability. In the first step genotype probabilities at a putative QTL are determined for individual animals using all phenotypic data. Genotype probabilities based on an animal's own phenotype are derived using a mixture distribution defined by the initial estimates of the QTL genotype effects and the initial estimate for the gene frequency in the total population. When adding information on all relatives an initial estimate of the gene frequency in the founder population is required. In FINDGENE the estimate for the total population is used.
In the second step a mixed linear model is used to set up a regression of phenotype on independent variables which will include the genotype probabilities, to obtain estimates of single QTL genotype effects, residual polygenic breeding value and other fixed environmental effects.
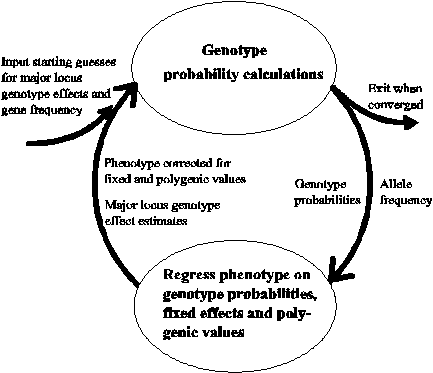
This is an illustration of the method used to arrive at converged estimates of major gene effects, b1 and b2, and calculate genotype probabilities for individuals. Genotype probabilities are calculated following Kerr and Kinghorn (1996), and gene frequency estimated by appropriate averaging of these probabilities. These probabilities are then fitted in a regression of phenotype on genotype probabilities and animal breeding values as described later. This regression yields estimates of breeding value and, after appropriate transformation, new estimates of b1 and b2. Phenotypes (P) are corrected for estimated breeding values ( ) in an attempt to reduce the influence of polygenic effects on the next calculation of genotype probabilities. The cycle illustrated is repeated sufficient times to give convergence in estimates of b1 and b2.
The step which calculates genotype probabilities is usually referred to as the GENEPROB step, named after the subroutine GENEPROB. The other step is usually referred to as the regression step.
A note on the implicit animal model.
If readers are familiar with programs such as PEST or DFREML they are aware that they have the capability to cater for almost any model desired by the user. The implicit animal model (IAM) is somewhat restricted in how it can handle the multiplicity of models that would be encountered in animal breeding. There is a restricted range of fixed effects, covariables and extra random effects allowable. However, this range should be able to accommodate almost any combination of effects. Please contact the authors if this is not the case. Currently the fixed effects accommodated are:
If your data set has more fixed effects, then the likely strategy is to nest these extra effects into the contemporary groups.
Currently the random effects accommodated are:
Information required for a FINDGENE analysis:
Pedigree information in a pedigree file:
This should be an ASCII file with fields of fixed length containing individual identity, sire identity and dam identity. Alphanumerics are permitted. It can be the same file as that at 4. below, including data following these three fields (but see ‘Missing records’).
This should be an ASCII file with fields of fixed length containing individual identity, plus any fixed effect classes, covariables and traits to be fitted. Identifications and fixed effects are defined as characters, and the covariate and observations as real numbers.
Missing values. The data file must NOT contain animals with missing observations. A zero entry will be interpreted as a valid observation. An animal with no observation will only have an entry in the pedigree file. This is the most effective way of dealing with missing observations.
You should provide, if possible, a statement declaring the model which you feel is most appropriate, especially in relation to fixed effects, covariates, maternal effects and common litter effects.
Results from FINDGENE analysis can be delivered to you together with some diagnostic software to aid interpretation and action planning.
Kerr, R.J. and Kinghorn, B.P. (1996). An efficient algorithm for segregation analysis in large populations. J. Anim. Breed. Genet. 113:457-469.
Kinghorn, B.P., Kennedy, B.W. and Smith, C. (1993) A method of screening for genes of major effect. Genetics 134, 351-360.
van Arendonk, J.A.M., Smith, C. and Kennedy, B.W. (1989) Method to estimate genotype probabilities at individual loci in farm livestock. Theoretical and Applied Genetics 78, 735-740.